| This page details the steps I followed to
re-align my Whole
Genome Sequencing (WGS) I ordered from Dante Labs into
format better suited for processing by YFull. NOTE: Much of the relevant and useful advise for the procedures followed on this page come from: |
||
| After the Dante Labs DNA sequencing was
complete, I downloaded all of the available files, but the
ones of the most value and interest were these: -r--r--r-- 1 darren darren 49967793271 Jan 25 05:45 xxx.bam -r--r--r-- 1 darren darren 8887488 Jan 24 08:44 xxx.bam.bai -rw-r--r-- 1 darren darren 29787997552 Mar 19 06:11 xxx_SA_L001_R1_001.fastq.gz -rw-r--r-- 1 darren darren 29787997552 Mar 27 21:18 xxx_SA_L001_R2_001.fastq.gz Unfortunately, as I was to determine later, my 'R2' fastq file was improperly compressed by Dante Labs, and there was a delay while I requested the file to be re-created properly. The easy way to test this success eventually was this way:
Now that I had FASTQ files, I opted
to process them through $ apt install fastq $ time fastp -i xxx_SA_L001_R1_001.fastq.gz -I xxx_SA_L001_R2_001.fastq.gz -o out.R1.fq.gz -O out.R2.fq.gz Read1 before filtering: total reads: 374028594 total bases: 55498491846 Q20 bases: 53576268480(96.5364%) Q30 bases: 50562803705(91.1066%) Read1 after filtering: total reads: 371183026 total bases: 55099763618 Q20 bases: 53370656021(96.8619%) Q30 bases: 50412293351(91.4928%) Read2 before filtering: total reads: 374028594 total bases: 55498491846 Q20 bases: 53576268480(96.5364%) Q30 bases: 50562803705(91.1066%) Read2 aftering filtering: total reads: 371183026 total bases: 55099763618 Q20 bases: 53370656021(96.8619%) Q30 bases: 50412293351(91.4928%) Filtering result: reads passed filter: 742366052 reads failed due to low quality: 5404220 reads failed due to too many N: 33930 reads failed due to too short: 252986 reads with adapter trimmed: 11926 bases trimmed due to adapters: 920082 Duplication rate: 22.3908% Insert size peak (evaluated by paired-end reads): 35 JSON report: fastp.json HTML report: fastp.html fastp -i xxx_SA_L001_R1_001.fastq.gz -I xxx_SA_L001_R2_001.fastq.gz -o out.R1.fq.gz -O out.R2.fq.gz fastp v0.19.6, time used: 8342 seconds real 139m1.732s user 417m34.937s sys 7m1.393s The generated output HTML page can be seen here and the JSON data can be found here. |
||
| From comments online, I suspected that the
BAM file created by Dante Labs was in an older format
known as hg19/GRCh37.
In order to verify this, I ran the following: $ samtools view -H xxx.bam This produced a report with references to 'grch37': ... @PG ID: Hash Table Build VN: 01.003.044.3.4.11-hv-7 CL: dragen --build-hash-table true --ht-reference /staging/human_g1k_v37_decoy.fasta --output-directory /staging/grch37/ --enable-cnv true --enable-rna true DS: digest_type: 1 digest: 0xF2543D4A ref_digest: 0xC2311E75 ref_index_digest: 0xB4307AF0 hash_digest: 0x7BF2A3E5 @PG ID: DRAGEN SW build VN: 05.021.408.3.4.11 CL: /opt/edico/bin/dragen -f -r /references/grch37 --sv-reference /references/grch37.fasta -1 /ephemeral/xxx/xxx_SA_L001_R1_001.fastq.gz -2 /ephemeral/xxx/xxx_SA_L001_R2_001.fastq.gz --output-directory /ephemeral/xxx --vc-sample-name xxx... ... |
||
| In order to get maximal accuracy and value
from what I eventually intended to send to YFull, I had to create
a new BAM file aligned with the newest standard known as hg38/GRCh38p13.
I could either upload my large FASTQ files to various
conversion services (for a small cost), or attempt to use
my moderately-powerful Linux desktop to do this on my own
(with free open-source tools). I opted for the
latter :) |
||
| The first step in the process of making my
own alignment was to download the reference assembly and
index it. I got the file using instructions here
and using the link here.
This is the command that I used: $ time bwa index GRCh38.p13.genome.fa.gz [bwa_index] Pack FASTA... 47.58 sec [bwa_index] Construct BWT for the packed sequence... [BWTIncCreate] textLength=6534235976, availableWord=471772820 [BWTIncConstructFromPacked] 10 iterations done. 99999992 characters processed. [BWTIncConstructFromPacked] 20 iterations done. 199999992 characters processed. [BWTIncConstructFromPacked] 30 iterations done. 299999992 characters processed. ... [BWTIncConstructFromPacked] 690 iterations done. 6374612482 characters processed. [BWTIncConstructFromPacked] 700 iterations done. 6398716386 characters processed. [BWTIncConstructFromPacked] 710 iterations done. 6418572210 characters processed. [bwt_gen] Finished constructing BWT in 710 iterations. [bwa_index] 2418.96 seconds elapse. [bwa_index] Update BWT... 14.75 sec [bwa_index] Pack forward-only FASTA... 19.15 sec [bwa_index] Construct SA from BWT and Occ... 819.65 sec [main] Version: 0.7.17-r1188 [main] CMD: bwa index hg38.fa [main] Real time: 3477.408 sec; CPU: 3299.025 sec real 57m57.638s user 54m46.199s sys 0m12.827s |
||
| The next step was to install the newest
version of the 'bwa' and 'samtools' utilities. For
'bwa', using LMDE4 (Debian 10 "Buster") this was as easy
as: $ apt install bwa For 'samtools', getting the source from GitHub and compiling it was the only option. Some preliminaries were needed first: $ apt install zlib1g-dev libbz2-dev liblzma-dev autoconf libncurses5-dev Next, I had to install hstlib-develop: - Get 'htslib-develop' from https://github.com/samtools/htslib - Unzip it - CD into the unzipped directory - Then: autoconf ./configure make sudo make install Then I had to install samtools-develop: - Get 'samtools-develop' from https://github.com/samtools/samtools - Unzip it - CD into the unzipped directory - Then: autoconf ./configure make sudo make install |
||
| Actually re-aligning my FASTQ files into a
new BAM file was a very resource-intensive process.
I determined that the 8gb of RAM in my desktop was not
sufficient, so I ordered, waited, and installed 16gb as a
replacement (I later reduced my installed memory to 12gb,
since there were suspicions that my machine could not
properly handle this -- despite the fact that none of the
memory tests that I ran revealed any problems). During most of the actual run, over 10gb of RAM was used, 44gb of temporary BAM files were produced (352 files!), and the 4 CPU cores in my i7-4600U Dell Chromebox were running full-tilt. Here is what 'top' looked like while I was doing this: top - 15:41:30 up 1 day, 41 min, 0 users, load average: 4.24, 4.20, 4.18 Tasks: 122 total, 1 running, 121 sleeping, 0 stopped, 0 zombie %Cpu(s): 98.3 us, 1.4 sy, 0.0 ni, 0.2 id, 0.1 wa, 0.0 hi, 0.0 si, 0.0 st MiB Mem : 11950.9 total, 167.8 free, 9384.9 used, 2398.2 buff/cache MiB Swap: 0.0 total, 0.0 free, 0.0 used. 2232.7 avail Mem
So this took 32.8 hours on my
relatively fast machine (!), and produced a new
GRCh38-aligned BAM file:
|
||
| Due to failed previous attempts, I was
careful to test the integrity of the newly-generated BAM
file: $ time samtools quickcheck xxx_darren_enns_grch38.bam real 0m0.476s user 0m0.005s sys 0m0.000s $ time zcat xxx_darren_enns_grch38.bam > /dev/null real 20m38.380s user 18m49.310s sys 0m15.023s |
||
I then needed to create an index file for
the new BAM file:
This produced an index for the new GRCh38-aligned
BAM file, which I renamed so that I could also use a
different utility (bamtools) to also create an index file
for comparison:
|
||
| At this point I had a newly-aligned BAM
file that could (?) have been submitted to YFull, but the
suggestion was to submit a greatly-reduced BAM file
containing only Y and MT chromosome data. This new
smaller BAM file was created this way: $ time samtools view -h -b xxx_darren_enns_grch38.bam chrY chrM > xxx_darren_enns_grch38.chrY.chrM.bam real 0m55.136s user 0m51.998s sys 0m0.945s This created the following file: -rw-r--r-- 1 darren darren 272344643 Mar 30 10:09 xxx_darren_enns_grch38.chrY.chrM.bam This much smaller BAM file is the one that I submitted to YFull for further analysis. Initial assessment from them confirms that my paternal haplogroup is R-S16361, a subclade of the R-U106 Y-chromosome haplogroup. For more information on my genetic ancestry, see here. |
||
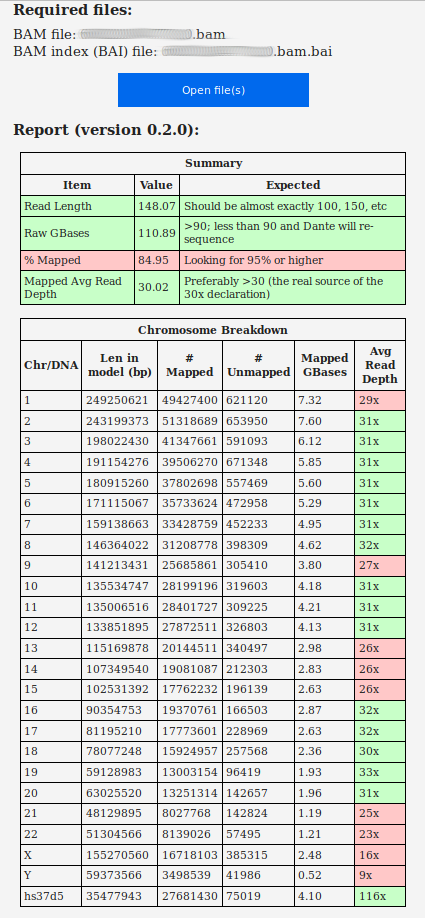
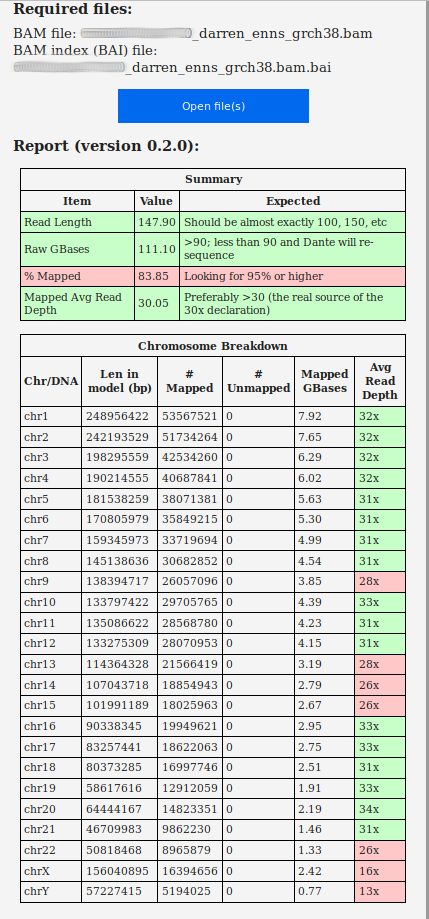
Quick and useful analysis reports of the
original (hg19) and newly-aligned (hg38) BAM files from qual.iobio.io:
|